如何高效通过国家实验室认可
2018-08-27
作者:
浏览数:2156
相信看到这篇文章的小伙伴都曾经历过(或将要经历)实验室资质认证认可的艰苦过程。小编曾经在检测机构供职长达5年之久,期间做过质量负责人、授权签字人和实验室主任,也经历过各种评审活动。实验室资质认可通常分为三个阶段:申请、评审和批准。小编认为这三个过程中申请最重要。俗话说的好“凡事预则立,不预则废”。申请准备的好不好直接关系到后面的两步是否顺利。今天,小编就跟大家聊一聊,申请的核心事情——验证报告的制作。
什么是验证报告?
简单粗暴的说验证报告就是给评审专家看的实验报告,目的是告诉评审组实验室已经有能力做相应的检测项目了。
实验室该如何准备验证报告?
1、实验前准备。拿到检测标准,仔细阅读检测标准,同时按标准要求,准备标准品、试剂、检测仪器或器具和样品。
2、定性。 (1)气相、液相: 在相同实验条件下进行样品测定,以检出的色谱峰的保留时间与标准样品相一致。检测顺序: 对照品(标准品),样品,样品加标,看是否保留时间一致。
(2)色谱-质谱:在相同实验条件下进行样品测定,以检出的色谱峰的保留时间与标准样品相一致,同时扣除背景后的样品质谱图中,所选择的离子必须都出现,而且所选择的离子丰度比与标准样品的离子丰度比相一致,则样品中含有该目标物。
定性要求有三个条件必须全部符合: a、保留时间偏差在2.5%以内;b、液质谱应有母离子,两个子离子。气质应至少2个定性离子。c、两个定性离子的丰度比与标品的丰度比在范围内,丰度在50%以上的,要求偏差在20%,丰度在50%以下时,偏差在50%以内。
(3)采用相对保留时间定性(待测物和基准物保留时间之比)。基准物必须是纯品,一般在各组分的保留值之间。
(4)根据色谱峰和特征光谱吸收峰结合法,避免误认相似保留时间内的杂质峰。
(5)和化学方法结合定性。
漂移的情况:在相同试验条件下,样品中待检测物质与同时检测的标准品具有相同的保留时间, 偏差在正负2.5%之内。a、 可能管路有泄漏;b、室温的变化造成的;c、流动相混合不均匀。
3、校准曲线。 (1)校准曲线包括工作曲线和标准曲线。 工作曲线是指曲线和样品的测定步骤完全一样,即需要预处理。标准曲线是指曲线与样品的测定步骤不一样,即不需要做预处理。校准曲线是消去了背景干扰得出来的。标准曲线是以标准溶液及介质组成的标准系列,标绘出来的曲线。
(2)校准曲线根据情况不同,分内标法或外标法。外标法在分光、吸光度、原子吸收、原子荧光、ICP、液相色谱应用多;内标法在气相、气质、液质中应用的较多。
(3)试验点数量的选则:原子光谱分析中,一般至少为3个点(不包括空白),色谱分析要求校准曲线至少5个点(不包括空白),可以每个浓度测定三次,取平均值。
(4)确定线性响应范围,并计算出回归方程和相关系数。
4、样品前处理。食品、化学品、饲料、肥料、电子、金属、玩具、纺织品、环境(水质、土壤、空气)的采样和样品制备,应参见相关的国标、行标和技术规范。
5、样品定量。仪器法:根据仪器响应值在标准曲线或工作曲线上,计算出浓度,从而计算出样品中的被测物含量。滴定法:根据滴定的体积,扣除空白值,计算出样品的被测物含量。如果用的是微量滴定管(精确到0.05ml),读数可以精确到小数点后3位(一般最后小数只估计到0或5,如9.850ml, 9.855ml),否则,只能到小数点后2位。重量法:应根据标准的要求,测出含量。如果标准需要恒重,烘箱烘干后,应有恒重的过程,满座恒重的条件,如前后两次称重的差值小于2mg(如GB5009.3食品中水分的测定),并记录数值。
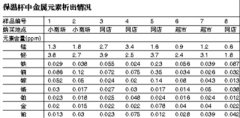
6、精密度实验。精密度一般是指在一定条件下对同一被测物多次测定的结果与平均值偏离的程度。精密度反映了随机误差的大小。通常用标准差(S)或相对标准偏差(RSD%)表示。有时用某一个浓度的同一样品或标品连续进样5次、6次做出的结果,得出RSD%来表示精密度,指的是该组分的仪器稳定性、仪器精密度。如果样品有值,精密度就是样品平行做六份测定,并计算RSD%。
(1)如果样品有值,精密度就是样品平行做六份测定,并计算RSD%。
(2)如果样品未检出,精密度就采用样品加标,平行重复做六份测定,并计算RSD%。加标值为2倍-5倍方法定量限(可以用2.5倍)或常见限量指标MRL任意选一点。对于水质、食品、土壤等领域中涉及的容量(滴定)法、分光光度法,也可以采取加标的形式。重量法测精密度必须采取有值的样品。
(3)对于某些环境样品(如空气的)测定痕量组分的,分析4~7个实验室空白加标平行,如设定每个中含有每种被分析物质的浓度建议为2-5μg/L。这个浓度可以在校准范围的中间点。
计算每个平行样中每种分析物的测定浓度,及所有平行样中每种分析物的平均浓度、每个分析物的平均准确度和每个分析物测定值的精密度(相对标准偏差,RSD%)。 对于每个分析物和替代物,用实际值的百分数表示的平均加标回收为70-130%,RSD应小于30%。
7、准确度(加标回收率)。食品中理化检验,加标量一般选择三个点,方法测定低限、最高残留限量MRL或常见限量指标或2-5倍方法测定低限、10倍方法测定低限共三个水平实验)。具体参见:GB/T 27404-2008 实验室质量控制规范 食品理化检测。农药残留检验的规定(NY/T 788-2004)。
8、方法检出限。 (1) 色谱法:用加标回收的最低点来计算S/N,三倍信噪比时为检出限)和定量低限(10倍信噪比)。最小被测组分浓度的多次测定,一般为10次,计算的标准偏差,以最小偏差的3倍计算仪器检出限,以10倍标准偏差计算定量限。检出限=S/b (b标准曲线斜率)。(2) 食品、水质和土壤涉及分光法、容量法和重量法的检出限,参考GB/T 5750.3-2006 水质分析质量控制。(3)食品领域,可以根据GB 5009.001-2003 食品卫生检验 总则。
9、异常数据处理。Dixon法(Q值检验法),是迪克森(W.J.Dixon)在1951年专为分析化学中少量观测次数(n<10)提出的一种简易判据式。当测量次数3~10次时,根据置信水平(如90%)确定可疑数据取舍。(1)将数据从小到大依次排列:X1,X2,X3,?,Xn-1,Xn,可疑数据将在开头(X1)或末尾(Xn)出现。(2)算出可疑数据与其邻近值之差的绝对值,即|X可疑-X邻近|。(3)虎出序列中最大值与最小值之差(极差),即Xn-X1。(4)用可疑值与邻近值之差的绝对值除以极差,所得的商称为舍弃商Q。公式为: Q=|X可疑-X邻近|/X最大-X最小》》进行实验室认可的意义!
查Q90%的临界值表,若计算所得Q值大于表中相应Q临界值,刚该可疑值应舍去,否则应被保留。
其他方法可参见GB5750.3-2006标准。
10、 测定下限(又叫定量限,定量下限)。分光光度法中按净吸光度0.02所对应的含量或质量浓度。或用色谱法中常用以10倍标准偏差(10N)计算定量限。
11、是否对方法偏离?如果是,对偏离进行说明。
12、结论。我公司对 ****中A、B含量 的检测符合******的要求。